





2026年4月27日,广州国家实验室胡文祥团队联合广州医科大学附属第一医院韩茜团队,在Nature Communications发表题为Dietary intake and BCAA metabolism regulate pulmonary fibrosis through KDM4A-mediated epigenetic remodeling in male mice的研究论文,综合运用转录组、代谢组、表观组、单细胞测序及体内外功能验证,系统揭示了支链氨基酸(Branched-Chain Amino Acids,BCAA)代谢重编程通过KDM4A-H3K36me3表观修饰轴调控肺纤维化的全新机制,并证实饮食限制BCAA、靶向抑制SLC7A5、增强BCAA分解代谢均可有效缓解肺纤维化,为IPF提供了新的代谢-表观调控靶点与可直接临床转化的干预策略。
特发性肺纤维化(Idiopathic pulmonary fibrosis, IPF)是一种进展性、致死性间质性肺疾病,以肺泡上皮反复损伤、肺泡成纤维细胞异常活化以及细胞外基质过度沉积为主要特征,患者肺功能持续不可逆下降,临床预后极差,现有临床一线药仅能减缓疾病进展,但无法逆转纤维化,且存在不良反应与费用昂贵等问题,亟须阐明新的致病机制并挖掘可转化的治疗靶点。代谢重编程是器官纤维化的核心驱动因素,糖酵解、脂肪酸代谢等通路已有大量研究,但支链氨基酸(亮氨酸、异亮氨酸、缬氨酸等)代谢在肺成纤维细胞活化及肺纤维化中的功能、调控机制及转化价值仍缺乏系统解析。
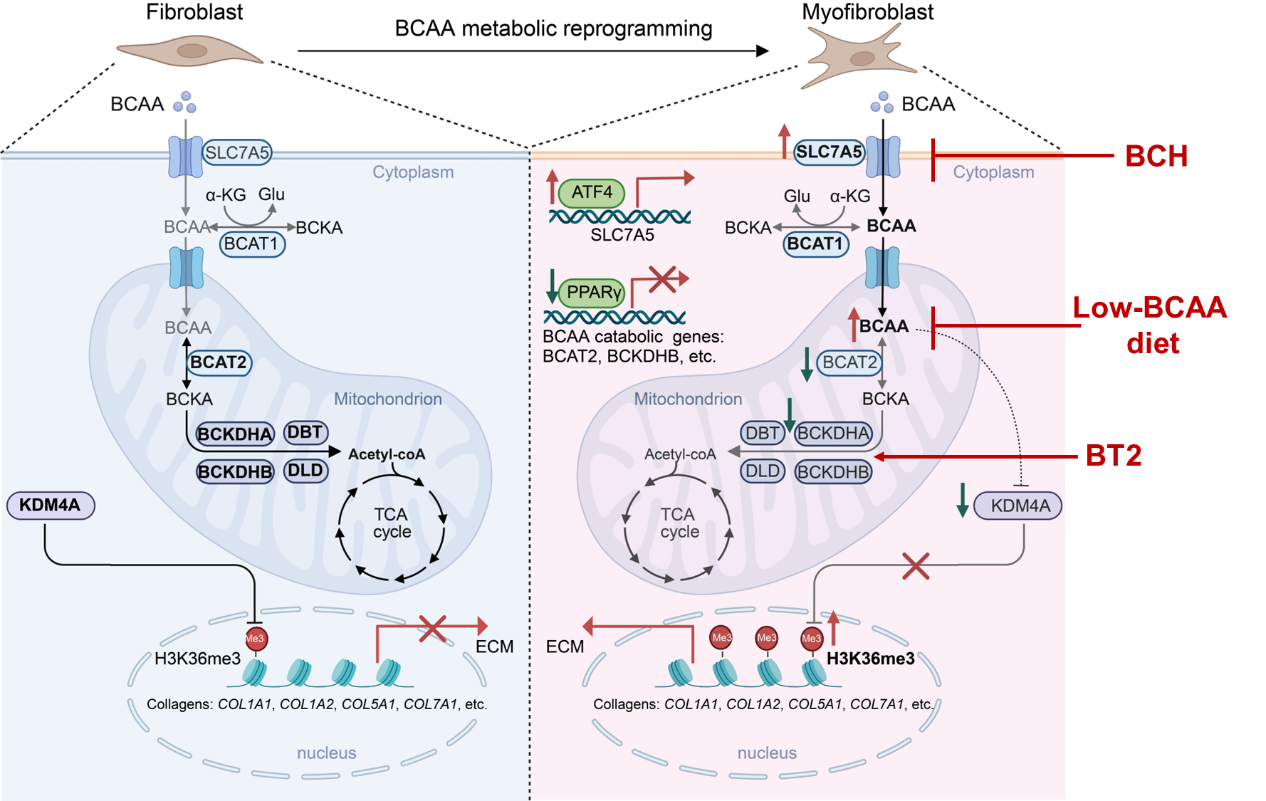
本研究以原代肺成纤维细胞、博来霉素(Bleomycin, BLM)诱导的小鼠肺纤维化模型以及IPF患者临床样本为研究对象,系统解析了BCAA摄取与分解代谢的重塑规律。研究人员首先对TGF-β1诱导的小鼠肺成纤维细胞和BLM诱导的小鼠纤维化肺组织进行多组学整合分析,发现BCAA合成与分解通路是纤维化过程中最显著富集的代谢通路,亮氨酸、异亮氨酸、缬氨酸在活化成纤维细胞和纤维化肺组织中显著累积;BCAA关键转运体SLC7A5显著上调,而BCAA分解代谢关键基因BCAT2、BCKDHA、DBT、DLD明显下调;13C-亮氨酸同位素示踪证实,纤维化成纤维细胞中BCAA分解代谢显著受损,表明BCAA摄取增强、分解受阻是肺纤维化中高度保守的核心代谢特征。
体内外功能回补与缺失实验明确证实,BCAA是调控肺纤维化胶原基因表达的关键代谢节点。BCAA回补可直接恢复BCAA缺失环境中成纤维细胞对TGF-β1的响应,显著恢复COL1A1、COL1A2和COL5A1等纤维化基因表达;小鼠饮水中添加BCAA可显著加重BLM诱导的肺组织胶原沉积与纤维化评分;BCAT2基因缺陷导致胞内BCAA异常累积,并显著加剧肺纤维化;单细胞测序显示,BCAT2缺陷小鼠中致病性成纤维细胞亚群显著增加,肺泡上皮细胞减少,炎症与纤维化通路全面激活。研究进一步鉴定出BCAA代谢紊乱的关键转录调控机制:PPARγ在纤维化刺激下表达下降,直接结合并抑制BCAA分解基因,罗格列酮激活PPARγ可恢复BCAA分解并抑制成纤维细胞活化;ATF4在肌成纤维细胞中显著高表达,并直接结合SLC7A5基因上促进BCAA摄取并驱动纤维化,揭示ATF4-SLC7A5轴与PPARγ-BCAT2轴协同驱动肺纤维化中BCAA异常累积。
研究团队发现,BCAA并非通过mTOR、AKT、TGF-SMAD等经典通路调控纤维化,而是通过表观遗传重编程发挥作用。BCAA缺失导致纤维化基因染色质开放程度显著下降,其中H3K36me3是表现出最显著BCAA依赖性的组蛋白修饰。筛选证实KDM4A是核心调控分子:BCAA累积使KDM4A下调,进而使胶原基因上H3K36me3显著富集并激活胶原基因表达。该研究首次提出并证实 BCAA-KDM4A-H3K36me3 表观调控轴是驱动肺纤维化的核心分子机制。
本研究同时提供了三种可直接向临床转化的干预策略:饮食限制BCAA,无论是预防性还是治疗性干预均可显著减轻肺纤维化,且效果呈剂量依赖性;SLC7A5特异性抑制剂BCH阻断BCAA摄取,可显著抑制成纤维细胞活化与肺纤维化;BCKDK抑制剂BT2可增强BCAA分解代谢,在预防与治疗小鼠模型中均显著缓解纤维化,疗效与临床用药尼达尼布相当。在IPF患者临床样本中,研究团队验证发现IPF肺组织中呈现SLC7A5上调、BCAT2下调的BCAA代谢紊乱特征,BCAA代谢基因表达与纤维化程度、肺功能(FVC、DLCO)显著相关,血清BCAA水平随病情加重显著降低,可作为IPF潜在预后标志物;BT2与BCH可直接抑制IPF患者原代成纤维细胞的纤维化表型。
特发性肺纤维化(Idiopathic pulmonary fibrosis, IPF)是一种进展性、致死性间质性肺疾病,以肺泡上皮反复损伤、肺泡成纤维细胞异常活化以及细胞外基质过度沉积为主要特征,患者肺功能持续不可逆下降,临床预后极差,现有临床一线药仅能减缓疾病进展,但无法逆转纤维化,且存在不良反应与费用昂贵等问题,亟须阐明新的致病机制并挖掘可转化的治疗靶点。代谢重编程是器官纤维化的核心驱动因素,糖酵解、脂肪酸代谢等通路已有大量研究,但支链氨基酸(亮氨酸、异亮氨酸、缬氨酸等)代谢在肺成纤维细胞活化及肺纤维化中的功能、调控机制及转化价值仍缺乏系统解析。
本研究以原代肺成纤维细胞、博来霉素(Bleomycin, BLM)诱导的小鼠肺纤维化模型以及IPF患者临床样本为研究对象,系统解析了BCAA摄取与分解代谢的重塑规律。研究人员首先对TGF-β1诱导的小鼠肺成纤维细胞和BLM诱导的小鼠纤维化肺组织进行多组学整合分析,发现BCAA合成与分解通路是纤维化过程中最显著富集的代谢通路,亮氨酸、异亮氨酸、缬氨酸在活化成纤维细胞和纤维化肺组织中显著累积;BCAA关键转运体SLC7A5显著上调,而BCAA分解代谢关键基因BCAT2、BCKDHA、DBT、DLD明显下调;13C-亮氨酸同位素示踪证实,纤维化成纤维细胞中BCAA分解代谢显著受损,表明BCAA摄取增强、分解受阻是肺纤维化中高度保守的核心代谢特征。
体内外功能回补与缺失实验明确证实,BCAA是调控肺纤维化胶原基因表达的关键代谢节点。BCAA回补可直接恢复BCAA缺失环境中成纤维细胞对TGF-β1的响应,显著恢复COL1A1、COL1A2和COL5A1等纤维化基因表达;小鼠饮水中添加BCAA可显著加重BLM诱导的肺组织胶原沉积与纤维化评分;BCAT2基因缺陷导致胞内BCAA异常累积,并显著加剧肺纤维化;单细胞测序显示,BCAT2缺陷小鼠中致病性成纤维细胞亚群显著增加,肺泡上皮细胞减少,炎症与纤维化通路全面激活。研究进一步鉴定出BCAA代谢紊乱的关键转录调控机制:PPARγ在纤维化刺激下表达下降,直接结合并抑制BCAA分解基因,罗格列酮激活PPARγ可恢复BCAA分解并抑制成纤维细胞活化;ATF4在肌成纤维细胞中显著高表达,并直接结合SLC7A5基因上促进BCAA摄取并驱动纤维化,揭示ATF4-SLC7A5轴与PPARγ-BCAT2轴协同驱动肺纤维化中BCAA异常累积。
研究团队发现,BCAA并非通过mTOR、AKT、TGF-SMAD等经典通路调控纤维化,而是通过表观遗传重编程发挥作用。BCAA缺失导致纤维化基因染色质开放程度显著下降,其中H3K36me3是表现出最显著BCAA依赖性的组蛋白修饰。筛选证实KDM4A是核心调控分子:BCAA累积使KDM4A下调,进而使胶原基因上H3K36me3显著富集并激活胶原基因表达。该研究首次提出并证实 BCAA-KDM4A-H3K36me3 表观调控轴是驱动肺纤维化的核心分子机制。
本研究同时提供了三种可直接向临床转化的干预策略:饮食限制BCAA,无论是预防性还是治疗性干预均可显著减轻肺纤维化,且效果呈剂量依赖性;SLC7A5特异性抑制剂BCH阻断BCAA摄取,可显著抑制成纤维细胞活化与肺纤维化;BCKDK抑制剂BT2可增强BCAA分解代谢,在预防与治疗小鼠模型中均显著缓解纤维化,疗效与临床用药尼达尼布相当。在IPF患者临床样本中,研究团队验证发现IPF肺组织中呈现SLC7A5上调、BCAT2下调的BCAA代谢紊乱特征,BCAA代谢基因表达与纤维化程度、肺功能(FVC、DLCO)显著相关,血清BCAA水平随病情加重显著降低,可作为IPF潜在预后标志物;BT2与BCH可直接抑制IPF患者原代成纤维细胞的纤维化表型。
图1 BCAA重编程调控肺成纤维细胞活化与纤维化示意图
广州国家实验室胡文祥研究员和广州医科大学附属第一医院韩茜主任医师为本文通讯作者。广州国家实验室助理研究员姚杰博士,中山大学博士研究生方苏和上海科技大学博士生雷妙为论文共同第一作者。本研究工作得到了广州国家实验室张炜研究员/苏金教授团队、中国科学院上海药物研究所王超群研究员、广州医科大学彭杨教授、南方医科大学荣知立教授和浙江大学饶威教授的大力支持。
论文链接:https://www.nature.com/articles/s41467-026-72273-3
论文链接:https://www.nature.com/articles/s41467-026-72273-3
